Aldeyra Therapeutics Achieves Primary Endpoint in Phase 3 Dry Eye Disease Clinical Trial of Reproxalap
New Drug Application Resubmission Anticipated in 2024
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20240808408806/en/
(Graphic:
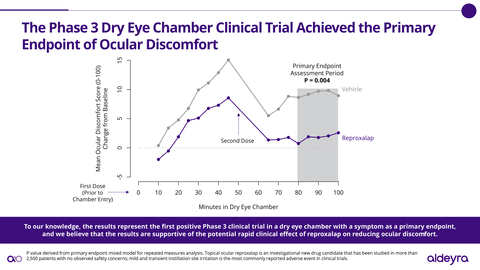
“To our knowledge, the results announced today represent the first positive Phase 3 clinical trial in a dry eye chamber with a symptom as a primary endpoint, and we believe that the results are supportive of the potential rapid clinical effect of reproxalap on reducing the ocular discomfort associated with dry eye disease,” stated
In the Phase 3 clinical trial, patients were administered vehicle (the drug product without the active ingredient) before and during exposure to a dry eye chamber in a manner that Aldeyra believes is consistent with the FDA’s dry eye disease draft guidance1. Qualifying patients were subsequently randomized to receive either reproxalap or vehicle before and during exposure to an additional dry eye chamber. Of the 132 patients randomized, 66 patients received reproxalap and 66 patients received vehicle. The primary endpoint was ocular discomfort, an FDA-accepted symptom of dry eye disease, from 80 to 100 minutes in the chamber. The dry eye chamber clinical trial was designed to satisfy the FDA’s New Drug Application (NDA) resubmission requirement, identified in the previously received complete response letter, of “at least one additional adequate and well-controlled study to demonstrate a positive effect on the treatment of ocular symptoms of dry eye.” Through the FDA Special Protocol Assessment process and additional comments, the FDA provided feedback on the clinical trial protocol and statistical plan.
To Aldeyra’s knowledge, in patients with dry eye disease, reproxalap is the first investigational drug with pivotal data supportive of acute and chronic activity in reducing symptoms, and the first investigational drug for chronic administration with pivotal data supportive of acute activity in reducing ocular redness. The potential NDA resubmission is anticipated in 2024. Based on FDA guidance, the resubmission NDA review period is expected to be six months.
There were no safety signals observed in the clinical trial, and reproxalap was observed to be well tolerated. Consistent with prior clinical trials, the most commonly reported adverse event was mild and transient instillation site discomfort. No treatment-related discontinuations were reported. Reproxalap has now been studied in over 2,500 patients.
Conference Call & Webcast Information
Aldeyra will host a conference call at
About Aldeyra
About Reproxalap
Reproxalap is an investigational new drug candidate in development for the treatment of dry eye disease and allergic conjunctivitis, two of the largest markets in ophthalmology. Reproxalap is a first-in-class small-molecule modulator of RASP, which are elevated in ocular and systemic inflammatory diseases. The mechanism of action of reproxalap has been supported by the demonstration of statistically significant and clinically relevant activity in multiple physiologically distinct late-phase clinical indications. Reproxalap has been studied in more than 2,500 patients with no observed safety concerns; mild and transient instillation site irritation is the most commonly reported adverse event in clinical trials.
Safe Harbor Statement
This release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995, including, but not limited to, statements regarding Aldeyra’s future expectations, plans, and prospects, including without limitation statements regarding: the goals, opportunity, and potential for reproxalap; the outcome and timing of the FDA’s review, acceptance and/or approval of a potential NDA resubmission for reproxalap and the adequacy of the data included in the original NDA and such NDA resubmission; and Aldeyra’s expectations regarding the labeling for reproxalap, if approved. Aldeyra intends such forward-looking statements to be covered by the safe harbor provisions for forward-looking statements contained in Section 21E of the Securities Exchange Act of 1934 and the Private Securities Litigation Reform Act of 1995. In some cases, you can identify forward-looking statements by terms such as, but not limited to, “may,” “might,” “will,” “objective,” “intend,” “should,” "could," “can,” “would,” “expect,” “believe,” “anticipate,” “project,” “on track,” “scheduled,” “target,” “design,” “estimate,” “predict,” “contemplates,” “likely,” “potential,” “continue,” “ongoing,” “aim,” “plan,” or the negative of these terms, and similar expressions intended to identify forward-looking statements. Such forward-looking statements are based upon current expectations that involve risks, changes in circumstances, assumptions, and uncertainties. Aldeyra is at an early stage of development and may not ever have any products that generate significant revenue. All of Aldeyra's development timelines may be subject to adjustment depending on recruitment rate, regulatory review, preclinical and clinical results, funding, and other factors that could delay the initiation, enrollment, or completion of clinical trials. Important factors that could cause actual results to differ materially from those reflected in Aldeyra's forward-looking statements include, among others, the timing of enrollment, commencement and completion of Aldeyra's clinical trials, the timing and success of preclinical studies and clinical trials conducted by Aldeyra and its development partners; delay in or failure to obtain regulatory approval of Aldeyra's product candidates, including as a result of the FDA not accepting Aldeyra’s regulatory filings, issuing a complete response letter, or requiring additional clinical trials or data prior to review or approval of such filings or in connection with resubmissions of such filings; the ability to maintain regulatory approval of Aldeyra's product candidates, and the labeling for any approved products; the risk that prior results, such as signals of safety, activity, or durability of effect, observed from preclinical or clinical trials, will not be replicated or will not continue in ongoing or future studies or clinical trials involving Aldeyra's product candidates in clinical trials focused on the same or different indications; the scope, progress, expansion, and costs of developing and commercializing Aldeyra's product candidates; uncertainty as to Aldeyra’s ability to commercialize (alone or with others) and obtain reimbursement for Aldeyra's product candidates following regulatory approval, if any; the size and growth of the potential markets and pricing for Aldeyra's product candidates and the ability to serve those markets; Aldeyra's expectations regarding Aldeyra's expenses and future revenue, the timing of future revenue, the sufficiency or use of Aldeyra's cash resources and needs for additional financing; the rate and degree of market acceptance of any of Aldeyra's product candidates; Aldeyra's expectations regarding competition; Aldeyra's anticipated growth strategies; Aldeyra's ability to attract or retain key personnel; Aldeyra’s commercialization, marketing and manufacturing capabilities and strategy; Aldeyra's ability to establish and maintain development partnerships; Aldeyra’s ability to successfully integrate acquisitions into its business; Aldeyra's expectations regarding federal, state, and foreign regulatory requirements; political, economic, legal, social, and health risks, public health measures, and war or other military actions, that may affect Aldeyra’s business or the global economy; regulatory developments in
In addition to the risks described above and in Aldeyra's other filings with the
1 www.fda.gov/media/144594/download
View source version on businesswire.com: https://www.businesswire.com/news/home/20240808408806/en/
Investor & Media:
Tel: (917) 618-2651
investorrelations@aldeyra.com
Source: